
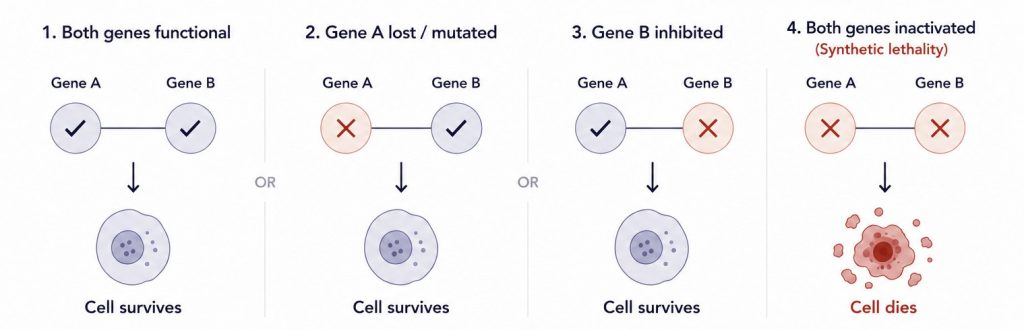
Two genes. Lose one in a cancer cell — it survives by adapting. Disable the second, and the cancer cell can no longer compensate.
That simple asymmetry lies at the heart of synthetic lethality, one of the most powerful and clinically promising ideas in modern oncology.
Unlike traditional drug targets, where you try to block something a cancer cell is doing too much of, synthetic lethality works by exploiting something a cancer cell has already lost. Tumors accumulate mutations. Those mutations close off certain survival pathways, and in doing so, make the cell entirely dependent on the ones that remain. Find the right dependency, pharmacologically block it, and the cancer cell collapses, while healthy cells, which still have both pathways intact, are spared.
The selectivity this creates is the basis of approved drugs already in use today. As computational approaches make it possible to map such interactions at scale, across thousands of gene pairs, across multiple tumor types, synthetic lethality is moving from a proof of concept into a systematic framework for drug discovery.
Cancer Therapeutics: How Cancer Mutations Become Therapeutic Vulnerabilities
In healthy cells, critical biological processes like DNA repair, cell division, and stress response are buffered by redundancy. Multiple pathways can accomplish the same task. Losing one gene is tolerable because another can compensate. This genetic buffering is what keeps normal cells stable even in the face of occasional damage.
Cancer disrupts that stability. As tumors evolve, they accumulate mutations that disable genes and silence pathways, often the very DNA repair mechanisms that would otherwise catch and correct further damage. This is what drives tumor progression, but it also creates a specific vulnerability: the cancer cell has traded away its backup systems and become dependent on what’s left.
This is where synthetic lethality becomes therapeutically relevant. If a tumor has already lost gene A through mutation or epigenetic silencing, its survival now depends on gene B carrying the full load. A drug that inhibits gene B not only slows the cancer; it removes the last functional repair route entirely. The cancer cell has nowhere left to go.
Healthy cells, which still retain gene A, tolerate the loss of gene B without consequence. That differential response, cancer cell death and healthy cell survival, is what makes synthetic lethality such a valuable framework for cancer treatment.
Proof of Concept: BRCA, PARP, and the Drugs That Validated the Idea
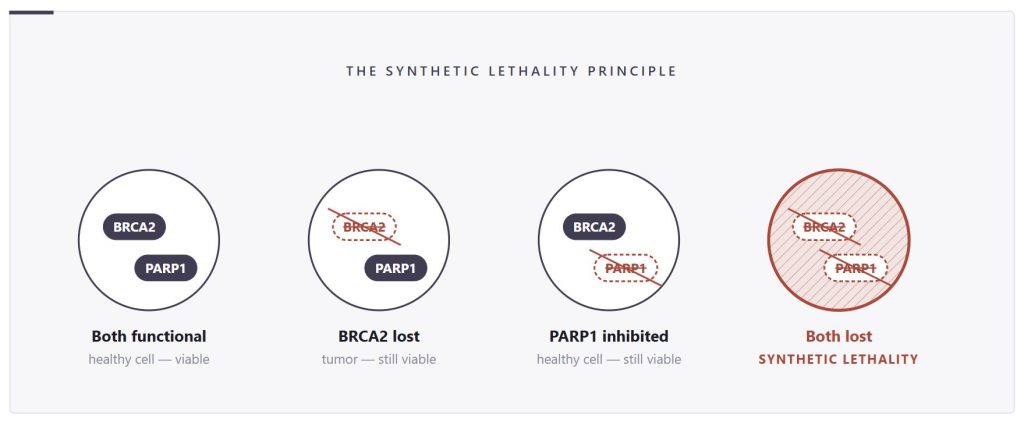
The clearest demonstration of synthetic lethality is the BRCA/PARP axis.
BRCA1 and BRCA2 are tumor suppressor genes that play a central role in homologous recombination repair, a high-fidelity mechanism that cells use to fix double-strand DNA breaks. In tumors where BRCA1 or BRCA2 is mutated or silenced, this repair pathway is compromised. These cells become dependent on an alternative mechanism called base excision repair, in which the enzyme PARP1 plays a critical role.
If you inhibit PARP in a BRCA-deficient tumor, you remove the last functioning DNA repair route. The cell accumulates irreparable DNA damage and dies. In a BRCA-intact healthy cell, homologous recombination compensates, and PARP inhibition is tolerable.
This interaction has now produced four approved drugs, olaparib, rucaparib, niraparib, and talazoparib, all of which are used clinically in prostate cancer patients with homologous recombination repair defects. What was once a theoretical genetic concept is now a standard-of-care treatment option, and its clinical validation has established tumor-specific vulnerabilities as actionable therapeutic targets.
The lesson is of strategic value: a synthetic lethal target identified computationally or experimentally can translate into an approved therapy. The question is how to find the next ones faster.
Beyond BRCA/PARP: An Expanding Frontier
The BRCA/PARP axis is the most clinically advanced example of synthetic lethal therapy, but it is far from the only one. Research is actively mapping new interactions, many of which are particularly relevant to prostate cancer:
- CDK12 loss and AR signaling: CDK12 is a kinase that regulates transcription-coupled DNA repair. Loss-of-function mutations in CDK12 are found in roughly 5–7% of metastatic castration-resistant prostate cancers. Emerging evidence suggests these tumors may be selectively vulnerable to inhibition of androgen receptor (AR) signaling, creating a synthetic lethal interaction that is currently under active clinical investigation.
- ARID1A and HDAC inhibition: ARID1A is a chromatin remodeling gene frequently mutated across multiple tumor types. Loss of ARID1A has been associated with synthetic lethality with inhibitors of HDAC6 and EZH2, both of which are druggable targets with existing compounds in development.
- WRN and microsatellite instability: Tumors with microsatellite instability (MSI), caused by defects in mismatch repair, have been shown to depend critically on the WRN helicase for DNA replication. This dependency represents a synthetic lethal vulnerability that is both mechanistically distinct from BRCA/PARP and potentially actionable with WRN inhibitors now entering early-phase trials.
The challenge is no longer proving that synthetic lethality works. It is identifying and validating new synthetic lethal interactions systematically across the vast genetic diversity of human cancers, as the number of potential genetic dependencies far exceeds what can be explored through experimental approaches alone.
The Discovery Problem: Why Most Synthetic Lethal Pairs Remain Unknown
The human genome contains approximately 20,000 protein-coding genes. The number of possible pairwise synthetic lethal interactions is, in theory, enormous, and the vast majority remain unmapped.
Experimental approaches to finding them, CRISPR screens, RNAi libraries, and genetic interaction mapping are powerful but resource intensive. Synthetic lethal screens are typically run in a specific cell line or tumor model, and results do not always generalize: a synthetic lethal pair that is active in one tumor subtype may be irrelevant or even absent in another. The biology is context-dependent in ways that make large-scale experimental coverage difficult and expensive.
There is also a drug development layer to the problem. Even when a synthetic lethal interaction is identified and validated experimentally, it is only therapeutically useful if one of the two genes is druggable: if a compound can be designed or repurposed to inhibit it effectively. Many identified pairs involve genes for which no tractable pharmacological strategy exists.
This is the gap that computational approaches are designed to close: not by replacing experimental validation, but by making the search space tractable. Prioritizing the gene pairs most likely to be both biologically active and pharmacologically actionable, in specific tumor contexts, before committing experimental resources.
Applying It to Prostate Cancer Research
Prostate cancer is a particularly compelling disease context for synthetic lethality-based drug discovery, for several reasons.
First, the prevalence of homologous recombination repair defects in advanced prostate cancer is high, estimated at 20–25% of metastatic castration-resistant cases, meaning a substantial patient population already carries the genomic vulnerability the BRCA/PARP framework exploits. The clinical validation of PARP inhibitors in this setting provides both a proof of concept and a baseline from which to identify further interactions.
Second, prostate cancer progression is marked by a well-characterized set of molecular events, AR amplification, PTEN loss, CDK12 mutation, and RB1 inactivation, each of which reshapes the cellular dependency landscape in ways that may create new synthetic lethal vulnerabilities not yet exploited therapeutically.
Third, the disease has a relatively high burden of treatment resistance. As tumors evolve under selective pressure from androgen deprivation therapy and second-generation AR pathway inhibitors, new molecular dependencies emerge. Mapping those dependencies computationally, and identifying compounds or combinations that target them selectively, is precisely the problem Hyper-C is built to address.
Project Objectives
The overall objective of Delta4’s PhD project within PROMOTE is to consolidate omics profiles in the context of prostate cancer to generate network-based molecular models for the disease. Network alignment methods shall be used to identify compounds that show beneficial interference between disease pathobiology and drug mechanism of action. A particular focus will be placed on compounds that affect target molecules involved in synthetic lethal interactions within the context of tumor progression. The potential of identified compounds and compound combinations targeting synthetic lethal interactions will be tested in combination with researchers from the Medical University of Innsbruck.
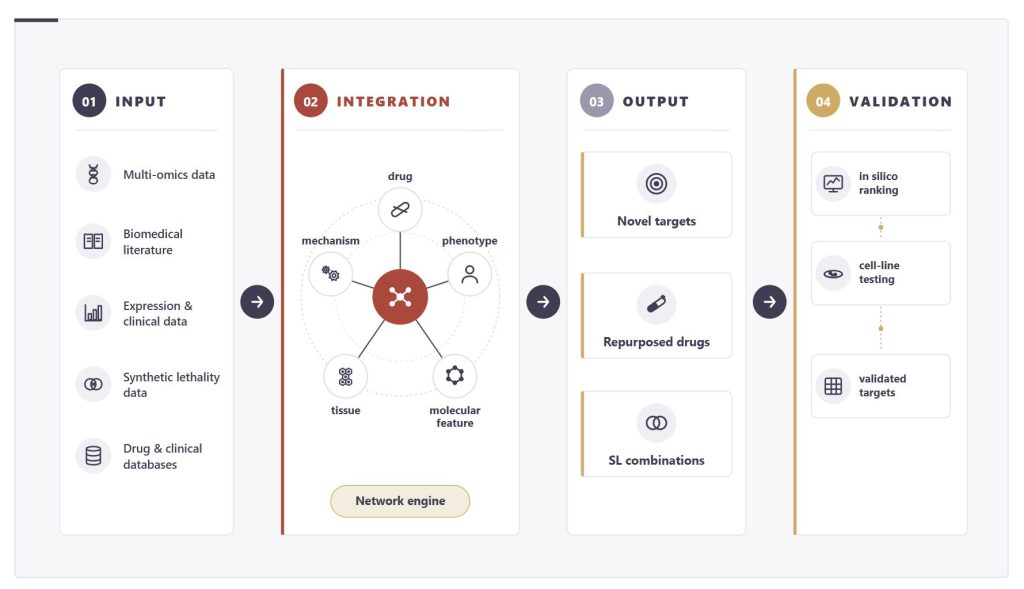
Delta4’s Approach: Computational Discovery at Scale
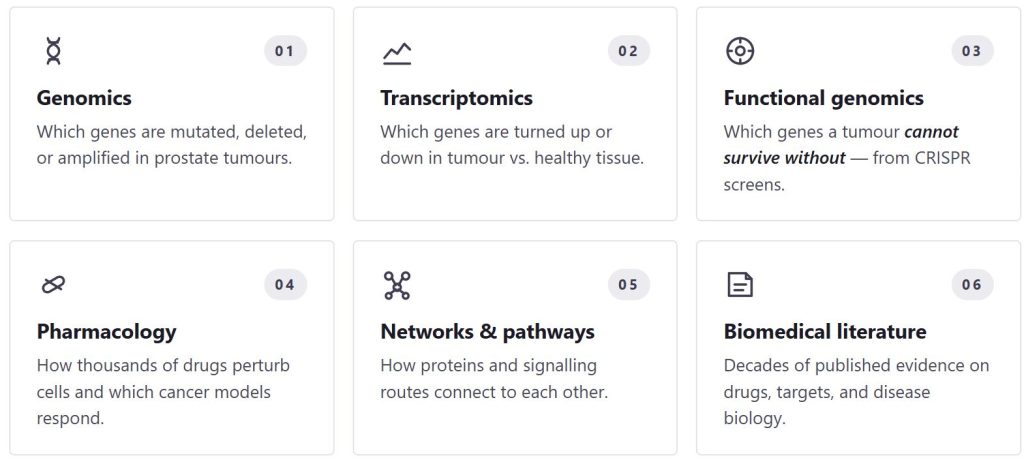
To find the right drug targets, we first need to understand the complex biology of prostate cancer at the molecular level — across many patients, many sample types, and many biological layers. We build that picture by integrating six complementary data types, each capturing a different facet of the disease:
Delta4’s Hyper-C platform models disease phenotypes as network responses to molecular perturbations. Rather than treating a tumor as a list of mutations or a drug as a single target, Hyper-C represents both as patterns of interconnected biological signals and identifies where those patterns intersect.
The engine is fed by an integrated multi-omic landscape built from heterogeneous public and PROMOTE’s consortium datasets, spanning six complementary data types:
- Genomic data — somatic mutations, copy number alterations, structural variants
- Transcriptomic data — tumor-versus-normal gene expression deregulation
- Functional genomics — CRISPR essentiality screens identifying gene dependencies in cancer cells
- Drug-target interaction data — known compound-protein binding relationships
- Pharmacological data — drug sensitivity profiles across cancer cell lines
- Biomedical literature — curated pathway and interaction databases
No single data source captures prostate cancer biology in full. The strength of the approach lies in integration: a target supported by tumor-versus-normal transcriptomic deregulation, recurrent genomic alteration, CRISPR-defined essentiality, and a known druggable interaction is a substantially stronger candidate than one supported by any single line of evidence.
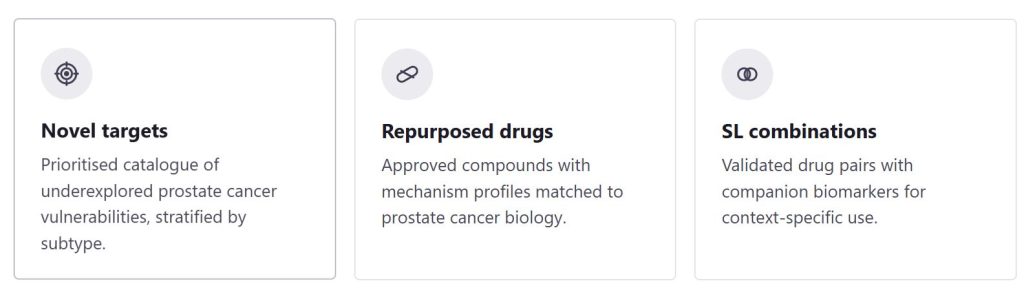
By aligning disease network states with drug mechanism-of-action profiles, Hyper-C identifies three categories of output:
- Novel drug targets that have not previously been associated with a given indication
- Existing drugs or clinical candidates with a mechanism that maps onto the disease network in an unexpected context
- Synthetic lethal drug combinations, where two agents targeting a synthetic lethal pair produce selective cancer cell death that neither achieves alone
For a deeper look at how multi-omic data integration works within this framework, see Delta4’s overview of multi-omics in drug repurposing. For the broader role of rational drug combination in modern medicine, see Delta4’s approach to drug combination screening.
What We Expect to Deliver
As a member of two EU Horizon research projects, including PROMOTE (PRostate cancer OMics Oriented inTErvention), Delta4 is particularly passionate about developing innovative therapies in this space. We are actively applying this methodology to prostate cancer as part of our current research program.
For more on Delta4’s development programs, including specific targets and therapeutic strategies currently under investigation, visit the development-programs page.
About the Author

Louiza Galou is an Application Scientist and Computational Biologist at Delta4, and an MSCA-funded PhD researcher within the EU Horizon Europe PROMOTE consortium. She holds a BSc in Bioinformatics from the University of Paris and an MSc in Bioinformatics and Engineering of Biological Platforms. Her research sits at the intersection of AI, genomics, and molecular medicine, with a focus on applying computational biology to decode the biological underpinnings of disease and advance precision therapeutics.
Prior to joining Delta4, Louiza worked at INSERM investigating structural variations in HIV-1 and HIV-2, at Owkin evaluating drug interactions at the transcriptomic level using omics data, and at Servier studying tumor heterogeneity and immunotherapy resistance mechanisms in non-small cell lung cancer.

